ความก้าวหน้าอย่างรวดเร็วของเทคโนโลยีการหาลําดับในช่วงสองทศวรรษที่ผ่านมาได้ให้ข้อมูลเชิงลึกที่ไม่เคยมีมาก่อนเกี่ยวกับโครงสร้างและหน้าที่ของจีโนมมนุษย์ แทนที่จะใช้เทคนิคความละเอียดต่ำที่ยุ่งยากมากขึ้นในการวิเคราะห์จีโนมมนุษย์ เช่น คาริโอไทป์และไมโครอาร์เรย์ ขณะนี้นักวิจัยสามารถใช้เทคโนโลยีการอ่านลําดับพันธุกรรมประสิทธิภาพสูงสําหรับการวิเคราะห์ตัวอย่างจีโนมมนุษย์ เพื่อค้นพบความแปรผัน (Variations) เชิงสาเหตุของโรคและความผิดปกติต่างๆ ที่ความละเอียดในระดับนิวคลีโอไทด์
โรคมะเร็งเป็นโรคที่มีซับซ้อนหลายด้าน ในปัจจุบันจำเป็นต้องใช้เทคนิคหลายอย่างเพื่อระบุลักษณะความแปรผันของจีโนมที่สามารถก่อให้เกิดหรือมีส่วนทำให้เกิดการลุกลามของมะเร็ง ด้วยเหตุนี้การศึกษาคุณลักษณะทางจีโนมโดยสมบูรณ์ (complete genome) ของตัวอย่างมะเร็งหนึ่งอาจทำได้ช้าและมีราคาค่อนข้างสูง ขณะที่เทคโนโลยี next-generation sequencing กลายมาเป็นกิจวัตรประจำวันมากขึ้นในห้องปฏิบัติการวิจัยโรคมะเร็ง แต่ก็ยังไม่สามารถระบุเครื่องหมายของจีโนมที่สำคัญจำนวนหนึ่งของโรคมะเร็งได้ เช่น SVs, DNA methylation และ transcript isoforms
“การอ่านลําดับด้วย Nanopore ให้การอ่านลำดับ Native DNA และการอ่านชิ้นส่วน RNA ที่มีความยาวการอ่านที่ไม่จํากัด การเรียกตัวแปร (variations) ที่แม่นยําพร้อมการวิเคราะห์ methylation ไปพร้อมกัน ผลลัพธ์แบบเรียลไทม์ และชุดอุปกรณ์ที่มีความยืดหยุ่นและราคาไม่แพง การอ่านลําดับด้วย Nanopore ให้ข้อมูลเชิงลึกที่ครอบคลุมที่สุดเกี่ยวกับจีโนมของมะเร็งโดยใช้เพียงเทคโนโลยีเดียว”
3 ความท้าทายของเทคโนโลยีอ่านลำดับแบบดั้งเดิมในงานวิจัยโรคมะเร็ง
|
Structural variants |
Methylation calling |
Transcript isoforms |
|
การอ่านลำดับแบบสายสั้น (short reads) ที่สร้างโดยเทคนิคแบบดั้งเดิมไม่สามารถครอบคลุมลำดับ structural variants หรือ repeat region ในบริเวณกว้างหรือที่มีความซับซ้อนได้ ซึ่งต้องใช้การวิเคราะห์เชิงคํานวณเพื่ออนุมานผลลัพธ์ เป็นผลให้ตัวแปรที่สําคัญหลายอย่างอาจผิดพลาด หรือตีความหมายผิดได้ |
เทคโนโลยีการอ่านลำดับแบบดั้งเดิมจำเป็นต้องอาศัยการทำ bisulfite conversion (ซึ่งเป็นเทคนิคการเตรียมตัวอย่างที่รุนแรง และลำบาก) เพื่ออนุมาน methylation ทางอ้อม ในกรณีที่ไม่มีลำดับอ้างอิง (reference sequence) ที่เหมาะสมอาจจำเป็นต้องมีการอ่านลำดับเพิ่มเติม ปัจจัยเหล่านี้สามาถเพิ่มความแปรผันของการทดลอง รวมถึงเวลา และค่าใช้จ่าย |
Transcript isoforms ที่แตกต่างกันสามารถเชื่อมโยงกับระยะของโรคที่แตกต่างกันได้ แต่การอ่านลำดับแบบสายสั้น (short reads) ที่สร้างโดยเทคนิค RNA-Seq แบบดั้งเดิมจะครอบคลุมความยาวของ transcript เพียงบางส่วนเท่านั้น ทำให้ยากต่อการประกอบและหาปริมาณ transcript isoforms อย่างแม่นยำ - ทำให้ได้ผลลัพธ์ของการแสดงออกของยีนที่ไม่สมบูรณ์ |
ความยาวในการอ่านไม่จำกัด
เทคโนโลยีการหาลำดับแบบดั้งเดิมนั้นอาศัยการแยกส่วน (fragmentation) และต่อด้วยการเพิ่มปริมาณ (amplification) ของชิ้น DNA หรือ RNA ซึ่งโดยทั่วไปจะมีความยาวระหว่าง 150 ถึง 300 bp ในระหว่างกระบวนการนี้ ข้อมูลตำแหน่งของแต่ละ fragment อาจขาดหายไป โดยงานส่วนใหญ่จำเป็นต้องประกอบชิ้นส่วนเหล่านี้กลับเข้าด้วยกันผ่านการทับซ้อนกับชิ้นส่วนอื่นๆ ซึ่งทำให้การวิเคราะห์ใช้เวลา และความซับซ้อนมากขึ้น แม้ว่าอาจไม่ใช่ปัญหาสำคัญมากนักเมื่อศึกษาเพียง SNPs หรือ mutation hotspots แต่อาจสร้างความสับสนให้กับการวิเคราะห์รูปแบบอื่นๆ ของความแปรผันของจีโนม เช่น Structural variants (SVs), repeat regions, phasing และ transcript isoforms (รูปที่ 1)
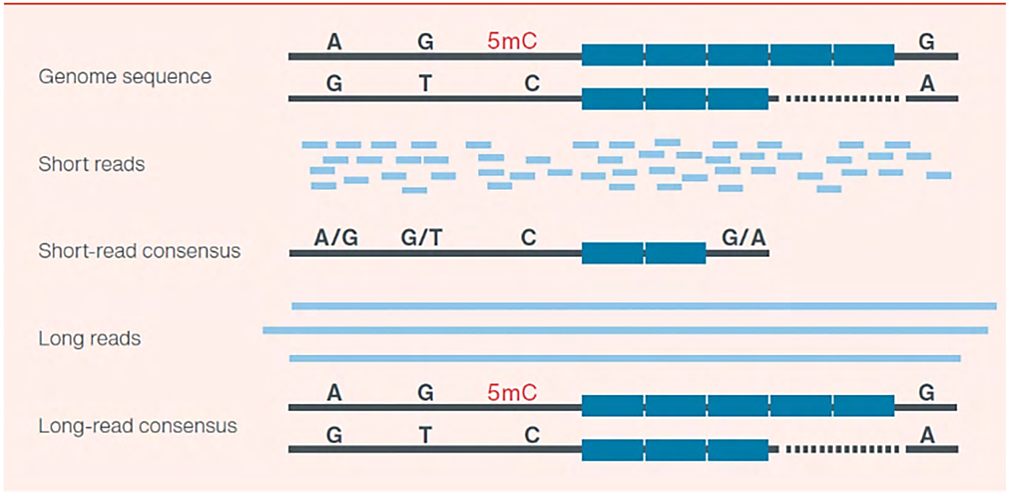
รูปที่ 1 แผนผังแสดงข้อดีของการอ่านลำดับโดยตรงแบบสายยาว สำหรับการประกอบแบบ De novo และการระบุความแปรผันของจีโนมต่างๆ รวมไปด้วย SNPs (อักษรสีดำ), repetitive regions (กล่องสีฟ้า) และ base modifications (อักษรสีแดง) การอ่านลำดับแบบสายยาว (Long reads) และแบบ Native DNA ให้ข้อมูลที่สามารถครอบคลุมพื้นที่จีโนมได้อย่างสมบูรณ์ รวมถึงข้อมูลการดัดแปลงเบส (base modifications) สำหรับ alleles ของทั้งมารดาและบิดา ในขณะที่การอ่านลำดับเบสแบบสายสั้น (short reads) นั้นก่อให้เกิดปัญหาในการศึกษา genomics repeats และ phasing ของจีโนม อีกทั้ง base modifications จำเป็นต้องมีการอ่านลำดับแยกต่างหาก
ด้วย Nanopore sequencing ความยาวในการอ่านจะเท่ากับความยาวชิ้นส่วนของ DNA หรือ RNA ทำให้สามารถวิเคราะห์ความยาวใดๆ ของโมเลกุล DNA หรือ RNA ได้ตั้งแต่โมเลกุลแบบสั้น เพื่อรองรับการใช้งานต่างๆ เช่น การวิเคราะห์ cell-free DNA แบบ ไปจนถึงโมเลกุลที่ยาวเป็นพิเศษ ซึ่งความยาวการอ่านมาก 4 ล้านเบส
“Multi-modality ของ Nanopore เป็นเทคโนโลยีในการวิจัย – DNA methylation, structural variation (SV), gene fusions, single mutations และ phasing – ที่ให้ข้อมูลครอบคลุมทั้งหมดในการทดสอบครั้งเดียว”
ที่มา: https://www.biodesign.co.th/service/catalogdownload_detail/22
